
The Nishida team from Kyushu University in Japan has revealed the molecular mechanism by which glutathione oxidized form can prevent ischemic heart failure.
Introduction
Mitochondrial dysfunction is a core feature in the occurrence and development of heart failure.
The excessive mitochondrial division mediated by Drp1 is closely related to myocardial aging and chronic heart failure after myocardial infarction (MI).
Oxidative stress is believed to worsen the prognosis of ischemic heart disease.
Glutathione (GSH), as an endogenous antioxidant, has been the subject of many failed therapeutic approaches in clinical studies of heart failure, suggesting that the redox-related treatment concepts need to be reexamined.
Sulfur-rich compounds (such as cysteine disulfide) possess a unique chain-like sulfur structure and redox activity.
They are widely present in the mammalian body and their metabolic imbalance is associated with cardiac dysfunction.
The cysteine residue of Drp1 (human Cys644) can undergo polysulfidation modification and negatively regulate its activity.
However, the mechanism linking abnormal hyper-sulfidation metabolism with the activation of Drp1, as well as the differential roles of glutathione oxidized form (GSSG) and reduced form (GSH) in regulating this process, remain unclear.
These limitations hinder the optimization of therapeutic strategies for ischemic heart disease.
On January 2, 2025, the Motohiro Nishida team from Kyushu University in Japan published a research paper titled "Polysulfur-based bulking of dynamin-related protein 1 prevents ischemic sulfide catabolism and heart failure in mice" in the top international academic journal Nature Communications.
This study found that oxidized glutathione, rather than GSH, can block the hypoxia-induced superoxide decomposition metabolism and Drp1 de-disulfide modification by inducing S-glutathionylation at the Cys644 site of Drp1, thereby inhibiting excessive mitochondrial division and cardiac aging.
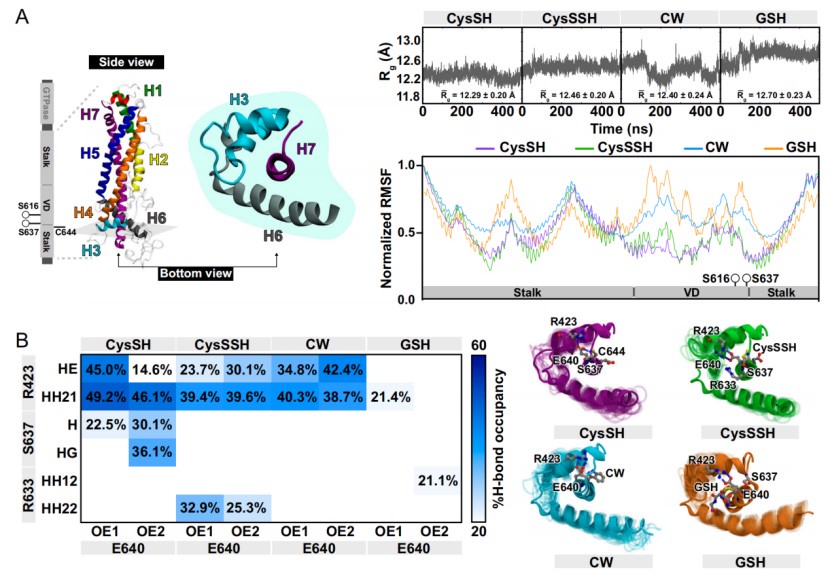
Molecular dynamics simulations and gene editing experiments have confirmed that the bulky modification at the Cys644 site reduces Drp1 activity by disrupting the interaction between Ser637-Glu640-Cys644. Drp1 C644S knock-in mice completely lose the cardiac protective effect of GSSG.
These findings have revealed a novel molecular mechanism by which oxidized glutathione can prevent ischemic heart failure, providing new targets and ideas for the treatment of related diseases.
Main Text
The researchers first discovered through a mouse voluntary movement model that the S-glutathionylation level of Drp1 in the cardiac tissue significantly increased after exercise, and it positively correlated with the GSSG content and the GSSG/GSH ratio.
In vitro experiments confirmed that the electrophilic forms of glutathione such as GSSG and S-nitrosoglutathione (GSNO) can induce the S-glutathionylation of Drp1, while GSH has no such effect.
Mass spectrometry analysis revealed that Cys644 and Cys470 of Drp1 are the main sites for GSSG-mediated S-glutathionylation.
Approximately 40% of the Cys644 sites have basic disulfide modifications (CysS-SH or CysS-SSH), and the disulfide-modified Cys644 is preferentially modified by GSSG to the S-glutathionylated form.
In neonatal rat cardiomyocytes, the interaction between Drp1 and S-glutathione can detected by proximity ligation assay after GSSG treatment, confirming the occurrence of Drp1 S-glutathioneylation within the cells.
To verify the protective effect of GSSG on cardiac cells, researchers found in the hypoxia model that GSSG pretreatment could significantly inhibit excessive mitochondrial division, improve the damage of mitochondrial cristae structure, and maintain mitochondrial membrane potential and oxygen consumption rate (OCR).
GSSG can reduce the proportion of SA-β-galactosidase-positive cells and p53-positive cells induced by hypoxia/reoxygenation (H/R), and improve the contractile dysfunction of cardiac muscle cells.
In human induced pluripotent stem cell-derived cardiomyocytes (hiPS-CMs), GSSG was also able to alleviate mitochondrial fragmentation and energy metabolism abnormalities induced by hypoxia, indicating that its protective effect is species-wide.
The mechanism exploration revealed that GSSG can inhibit the Drp1 GTP binding activity and particle formation induced by hypoxia through the Cys644 site.
Overexpression of glutathione reductase (Grx1) can partially reverse this inhibitory effect, confirming that S-glutathionylation is the key mechanism by which GSSG inhibits the activation of Drp1.
The sidestream smoke from cigarettes and environmental toxins such as methylmercury can induce pseudo-hypoxic stress similar to hypoxia, leading to the decomposition metabolism of sulfite and the de-sulfurization modification of Drp1.
GSSG treatment can effectively alleviate the mitochondrial dysfunction and cardiomyocyte aging caused by these toxins.
The researchers further verified in the mouse MI model that the level of hyperthiocyanate in the non-scarred myocardial area decreased while the level of hydrogen sulfide (H₂S) increased after MI, suggesting that the catabolic process of hyperthiocyanate enhanced.
Continuous infusion of GSSG (30 mg/kg/day) significantly increased the S-glutathionylation level of Drp1 in myocardial tissue, improved left ventricular ejection fraction (EF) and short-axis shortening rate (FS), and alleviated cardiac hypertrophy.
However, GSH and membrane permeability glutathione analogues (GEE) did not have such effects.
Electron microscopy analysis revealed that GSSG could alleviate mitochondrial fragmentation induced by MI, enhance the activity of mitochondrial complex I and the mtDNA/nDNA ratio, inhibit the reductive stress mediated by the increase in NADH/NAD⁺ ratio, but did not affect the level of oxidative stress marker 4-HNE.
The Drp1 C644S heterozygous knock-in mouse experiment confirmed that the mutant mice suffered more severe cardiac dysfunction after MI, and GSSG was unable to exert its protective effect.
It clearly demonstrated that the Cys644 site is a necessary site for the protective effect of GSSG on the heart.
Molecular dynamics simulations show that the polysulfide modification or S-glutathionylation modification at the Cys644 site increases spatial steric hindrance, disrupting the hydrogen bond interaction between Ser637 and Glu640, thereby reducing the activity of Drp1.
Moreover, the Drp1 E640A mutant can inhibit the activation of Drp1 induced by hypoxia and its interaction with the actin-binding protein FLNa.
Summary
This study systematically elucidated the molecular mechanism by which GSSG prevents ischemic heart failure:
Hypoxia or pseudo-hypoxia stress induces the decomposition metabolism of hyperthiols, resulting in the de-thiosylation modification and activation of the Cys644 site of Drp1, thereby triggering excessive mitochondrial division, myocardial aging and cardiac dysfunction.
GSSG can specifically induce S-glutamylation at the Cys644 site of Drp1.
Through bulky modification, it disrupts the interaction of Ser637-Glu640-Cys644, inhibiting the activation of Drp1 and the downstream pathological processes.
However, GSH does not have this effect.
This study for the first time revealed the functional differences between GSSG and GSH in ischemic heart disease, clarified the crucial role of the hyper-sulfuration modification and S-glutathionylation modification at the Drp1 Cys644 site in regulating mitochondrial dynamics, provided new therapeutic targets and strategies for ischemic heart failure, and has significant scientific value and clinical translational potential for optimizing redox-related therapies.
Paper link:
https://doi.org/10.1038/s41467-024-55661-5